Manual
ARCTIC-3D (ARCTIC-3D: Automatic Retrieval and ClusTering of Interfaces in Complexes from 3D structural information) is a software for data-mining and clustering of protein interface information.
In short, the software first retrieves all the available interface information for the selected protein, and then separates different interfaces according to a geometric measure and a clustering algorithm.
Input
- UNIPROT ID (mandatory): this is the unique identifier of your protein. If you don't know it, you can easily type the name of your protein in the UNIPROT database and check the resulting output. As an example, if you type Hemoglobin you can see that there are several proteins related to it, the first one being Hemoglobin subunit beta. The UNIPROT ID of such protein is the ENTRY string appearing on the left, in this case P68871
Once you know your UNIPROT ID, you can already submit an ARCTIC-3D run by simply pasting this string in the UniprotID field of the web interface:

Press Submit and wait a few seconds for the software to process your query!
Optional arguments
We saw the basic ARCTIC-3D usage, now let's see how you can tweak the software output with a few tunable parameters.
Interface parameters
- Interface file: in a standard ARCTIC-3D submission, the software queries the PDBe graph API to get all the interface information available for the input protein. But you may have your own set of interfaces! maybe coming from experiments or computational modelling. An example of interface is provided here
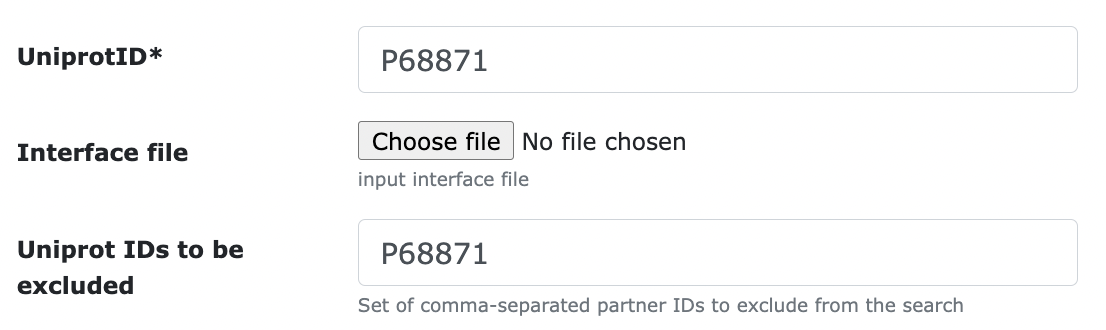
- Uniprot IDs to be excluded: you can fill in this field if you're not interested in some of the interfaces formed by your protein and don't want them to be considered. For example, to exclude all the homomeric interfaces of P68871:
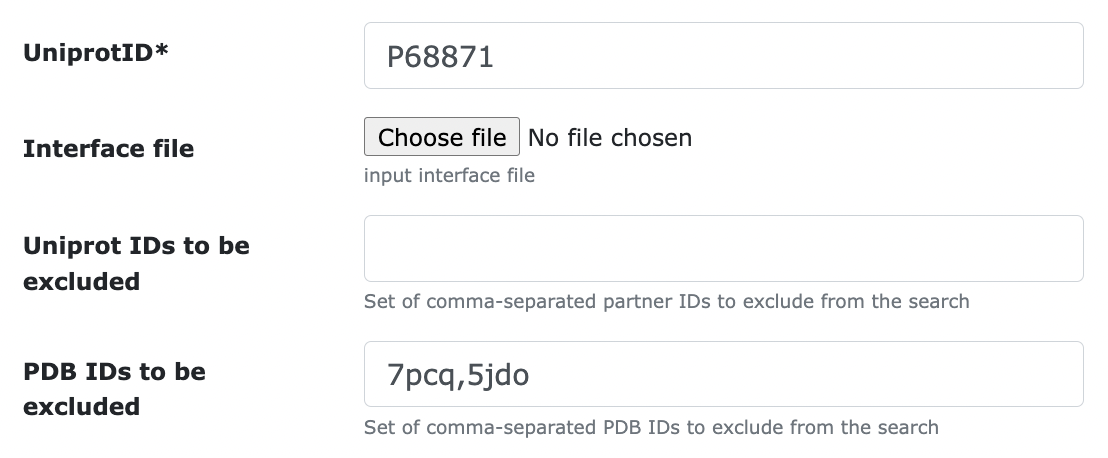
- PDB IDs to be excluded: sometimes you don't want a specific PDB file to be considered in the interface retrieval. For example, you don't trust that specific interface, or you aim at excluding it for benchmarking purposes. To exclude pdb files 7pcq and 5jdo from the search:
- Interface coverage cutoff: this parameter is used select the interface coverage, namely the fraction of residues that should be present in a given PDB file to retain the interface. Setting this value to 0.5 means that every retrieved interface should have at least 50% of the residues present in the PDB file.
Structure parameters
- PDB ID to be used: by default ARCTIC-3D downloads the PDB file that retains the highest number of interfaces, but you can specify which file you want to use.
- Chain ID to be used: you can also select which chain to use.
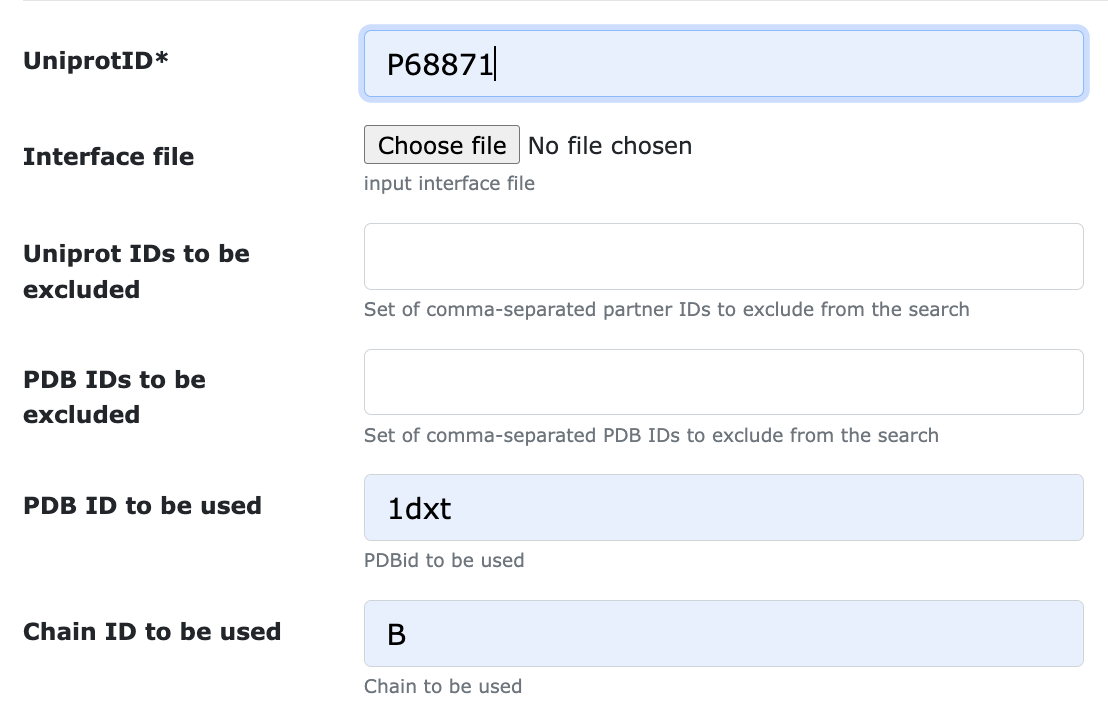
To force ARCTIC-3D to use PDB file 1dxt, chain B:
Clustering parameters
The aim of this parameter is to change the way the structural clustering of interfaces is performed. Detailed documentation about the hierarchical clustering algorithm is available here.
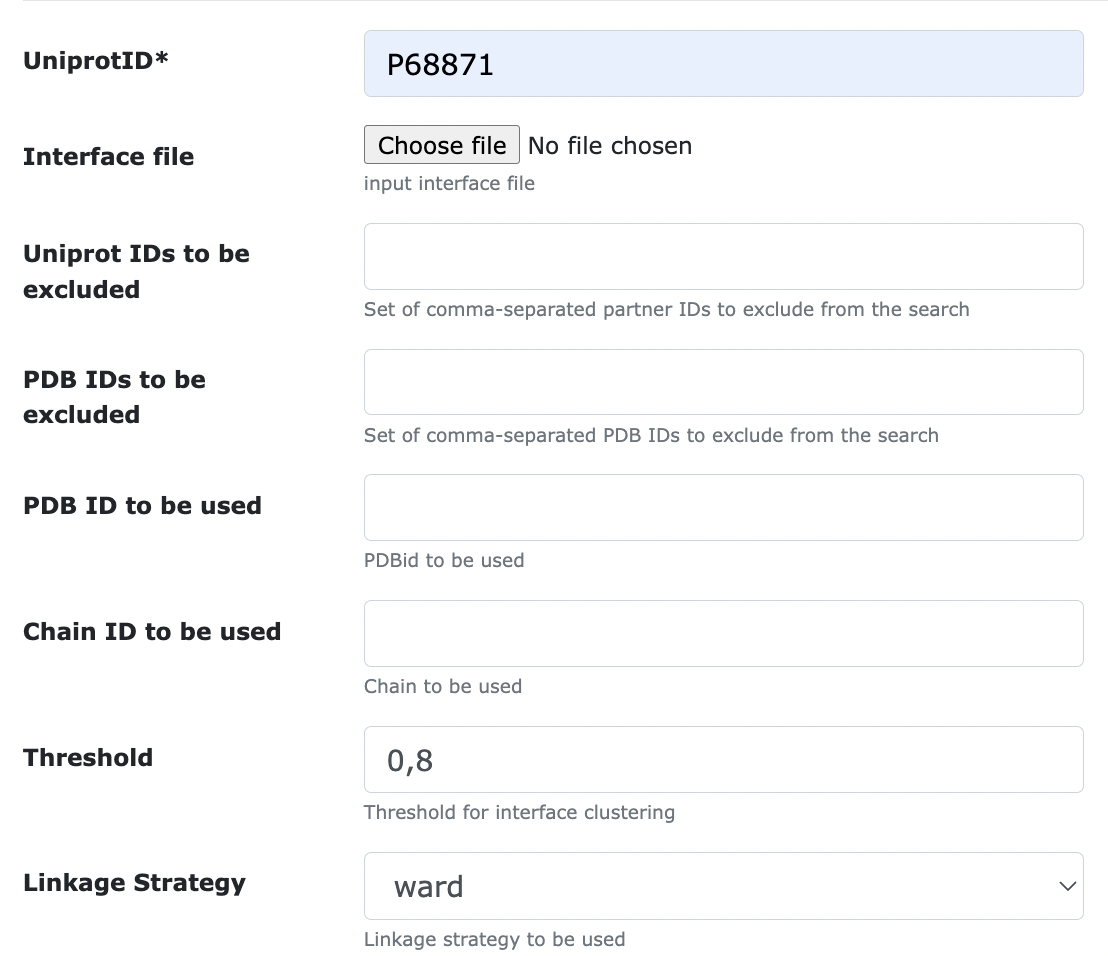
- Threshold : the cutoff distance to be used to cut the hierarchy of interfaces. In short, a lower number means that the clustering will be stricter, thus giving rise to more binding surfaces. Instead, a higher number corresponds to a looser clustering, with less, more heterogeneous surfaces in output. PS: the threshold value should be lower than 1.0;
- Linkage strategy : defines the way in which interface clusters should be grouped together. The default value for this is average, where the distance between two clusters is calculated as the average pairwise distance between their elements
- Minimum cluster size : there might be cases in which interface clusters consist of only one or two residues. The interfaces belonging to these clusters are typically not important from the biological point of view, as there's no specificity in a single amino acid. You can discard these "minimal" binding surfaces with this parameter. As an example, setting the Minimum cluster size to 3 will eliminate all clusters with one or two residues.
To cluster interfaces using the Ward linkage strategy and a threshold distance of 0.8:
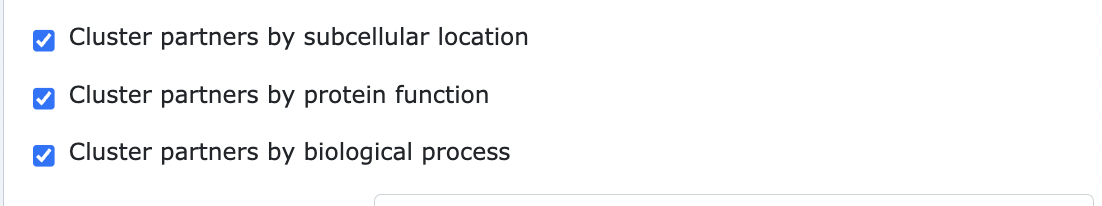
Clustering partners with QUICKGO annotations
When submitting an ARCTIC-3D run it is possible to cluster the binding partners of the input protein using the annotations provided by the QuickGO database. Partner proteins can be grouped based on their subcellular location, molecular function and biological process (an example available here).